A non-hypothesis driven discovery in science is exciting because it’s entirely unexpected. It is even more so when that discovery opens the door to new ideas that may save the lives of millions of people.
The Malaria parasite (Plasmodium sporozoite) infects approximately 200 million and kills over 400,000 people annually. We don’t produce effective antibodies to overcome this deadly infection. This is despite our ability to produce protective antibodies against a variety of pathogens by somatic hypermutation (SHM) and B cell selection.
Research on Malaria infection has identified an immunodominant region (where the immune system usually targets) on a parasite surface protein. This frequent target, PfCSP, is composed of amino acid repeats (Asparagine-Alanine-Asparagine-Proline, NANP38). Anti-NANP antibodies provide protection against parasite infection but attempted human vaccines have not been able to reliably generate these protective antibodies. They provide limited protection and the antibodies are short-lived.
That creates a question: why aren’t we capable of generating stable, protective anti-NANP antibodies more effectively? What are we missing?
To answer this question, researchers repeatedly exposed volunteers to high doses of the Malaria parasite. These volunteers took a course of chloroquine, a drug that prevents the full disease from developing, but still endured the sporozoite phase of the infection. Neutralizing antibodies were isolated from these volunteers to further understand the molecular details of their immune reactions.
Here, researchers selected five representative neutralizing antibodies for further analysis. They selected antibodies that were either immature (germline) or mutated by SHM. The first step was to determine the uniqueness and specificity of the two genes that encoded the variable domains that selected for anti-parasite activity. They mix-and-matched the heavy and light chains to uncover which antibody conferred protection and which did not.
If they replaced the light chain with one from a different gene or added an amino acid to the CDR3 region (replace KDCR3:8 region with a 9 amino acid CDR3), the antibodies lost their high-affinity to the parasite surface protein. If they made mutations to mimic highly similar heavy chain genes, binding affinity was reduced or eliminated. And when they tested these mutant antibodies to see if they protected liver cells from parasite infection, they failed. Altogether, these in vitro experiments validated the importance of selected antibody gene features in recognizing the antigen.
Sequence analysis of the variable domains revealed four SHM-driven mutations – three in the heavy chain ( H.Ser31, H.Val50, and H.Lys56) and one in the light chain (K.Asn93). When they reverted these mutations back to the germline amino acids and compared binding affinity to a short NANP3 peptide, it appeared only H.Ser31 and H.Val50 were important modifications.
This immediately raises the question: why were antibodies with H.Lys56 and K.Asn93 mutations selected for in B cell expansion? They have no (immediately) apparent value to the immune response.
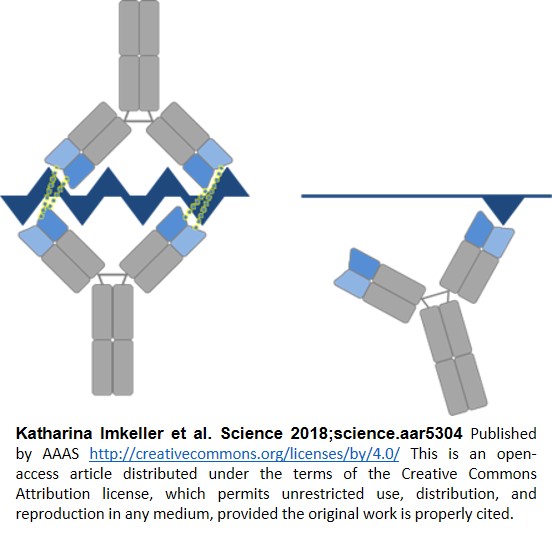
To investigate the specific roles of these mutations, the atomic-resolution structure of one of the antibodies with a longer 5-repeat NANP peptide was determined using protein crystallography methods. The structure revealed that two identical antibodies bound to the single peptide with each antibody binding to a separate but closely positioned NANP repeat. Even more surprisingly, the variable domains of the two antibodies directly interacted with each other in a head-to-head configuration (Fig. 1). With thousands of antibody structures published, seeing two human antibodies interact this way was entirely unique and unprecedented.

Proteins in a crystal are in a solid state, so conformations and interactions may or may not reflect the protein as it exists in a solution. To show this interaction is real, they mixed the antibody and full-length PfCSP with 38 NANP repeats in solution and found multiple antibodies bound to one PfCSP molecule in a head-to-head fashion (Fig.2).

Many amino acid contacts between adjacent variable domains were identified. In particular, H.Lys56 and K.Asn93 made direct contacts with the adjacent antibody. When they mutated these amino acids, antibody affinity to the longer NANP5 peptide was significantly reduced. So-H.Lys56 and K.Asn93 ARE critical mutations for antigen binding because they coordinate antibody-antibody interactions on closely positioned antigens.
More importantly, when they created antibodies with mutations that disrupted the head-to-head interaction, B cell activation was delayed. This described for the first time how affinity maturation by SHM can generate antibody-to-antibody (homotypic) interactions that help to enhance the immune response.
But is this just a single, isolated and fortuitous result? Or a recurring phenomenon? In response, researchers also analyzed a second population of selected human B cells with antibodies encoded by a different heavy chain gene. Antibodies produced by these cells bound to the NANP5 peptide in a different way, but SHM-driven mutations enhanced the same head-to-head interaction.
The discovery of antibodies that cooperate to improve an immune response was entirely unexpected. We now have a mechanistic explanation for why our immune system selects for these antibodies. And the authors go on to speculate that these types of head-to-head antibodies may occur in response to repetitive antigens in other pathogens as well (Fig. 3).
With a better understanding of the mechanistic processes underlying immunity from the Malaria parasite, researchers can engineer antibodies with these characteristics for direct antibody treatment. Even better, they can work to engineer preventative vaccines containing antigens that promote head-to-head neutralizing antibody production.
Reference:
Imkeller, K. et al. Antihomotypic affinity maturation improves human B cell responses against a repetitive epitope. Science 360, 1358-1362 (2018). doi: 10.1126/science.aar5304.
Steve Bryson has a background in drug- and vaccine-discovery with focus on structural biology. He loves creating content for science communications. Find him on Twitter @stevebrysonphd
